РЕАЛИЗАЦИЯ ИНТЕРАКТИВНОСТИ В МОЛЕКУЛЯРНОМ РЕДАКТОРЕ MOLRED
С.Н. ЖАБИН, В.Б. СУЛИМОВ
ООО «Димонта», Москва, Россия
Научно-исследовательский вычислительный центр Московского государственного университета имени М.В.Ломоносова, Россия
vladimir.sulimov@gmail.com (автор В.Б. Сулимов)
Оглавление
Аннотация
Описаны основные интерактивные возможности программы-редактора молекул MolRed и приведены особенности реализации этих возможностей. Поясняются способы формирования главной рабочей области программы в виде трёхмерной сцены, методы выделения атомов и молекул, методы редактирования геометрических и структурных параметров, в том числе особенности реализации функций undo и redo.
Ключевые слова: молекулярный редактор, молекулярное моделирование, компьютерная разработка лекарств, нанотехнология, молекулы, супрамолекулы, квантовые точки, докинг, SES – поверхность исключенного объема, триангулированная, интерактивность, щелчок мышью, виртуальная камера, OpenGL , 3D-сцена.
1. Введение
При разработке программ и программных комплексов для молекулярного моделирования, а также при их использовании необходимо иметь возможность наглядного представления молекулярных объектов. Область применения методов молекулярного моделирования непрерывно расширяется в связи с прогрессом в развитии технологий получения материалов с заданными свойствами. Так, в нанотехнологиях имеют дело с созданием новых веществ с заданной атомно-молекулярной структурой путем контролируемого манипулирования атомами и молекулами, и здесь использование методов молекулярного моделирования является принципиально необходимым. Другой перспективной областью применения методов молекулярного моделирования является разработка новых лекарств, где эти методы дают существенное повышение эффективности разработки новых лекарственных соединений на начальном этапе, когда конструируются молекулы-ингибиторы, избирательно связывающиеся с заданными белками-мишенями, определяющими развитие патологии (см., например, [1]). При использовании в этих областях методов молекулярного моделирования необходимо иметь возможность создавать, рассматривать, редактировать и измерять геометрические характеристики атомно-молекулярных объектов: молекул, супрамолекул, квантовых точек, нанокластеров и т.д. Такие системы с геометрической точки зрения представляют собой сложные структуры, и для их построения и анализа, а также анализа изменения этих структур, произведённых с помощью тех или иных программ молекулярного моделирования, необходим соответствующий инструмент. Молекулярный редактор и является таким инструментом. Более того, практика показала удобство использования молекулярного редактора для запуска различных программ молекулярного моделирования.
На данный момент существует множество программ, позволяющих просматривать и редактировать молекулы и их системы [2]. Как правило, такие программы являются закрытыми продуктами и не предоставляют возможности расширения их функциональности. Поэтому их интеграция вместе с разрабатываемыми в нашей лаборатории программами затруднена. Исключением является проект Avogadro [3], который представляет собой GNU -проект, разрабатываемый около двадцатью независимыми разработчиками. В рамках этого проекта интеграция наших программ возможна в виде «плагинов». Однако в проекте Avogadro , на наш взгляд, есть ряд недостатков, которые заставляют нас отказаться от его использования. Так, можно отметить практически невозможность работы с большими молекулами, такими как белки, поскольку при этом частота смены кадров становится очень низкой. Вообще проблема больших молекул свойственна многим существующим редакторам молекул.
Таким образом, создание своей программы – молекулярного редактора - с дальнейшей возможностью интеграции с имеющимися программами молекулярного моделирования представляется обоснованным.
В тоже время, несмотря на многообразие имеющихся программ для просмотра молекул, не удалось найти описания того, как эти программы сделаны. Зачастую упоминается лишь язык, на котором написана программа. Поэтому описание нашей программы весьма актуально, поскольку даёт представление об основных проблемах, решаемых при создании программы – редактора молекул.
В качестве оригинальных элементов, отличающих программу MolRed , можно выделить ряд характерных функциональных возможностей:
• возможность работы с молекулами, содержащими очень большое количество атомов – порядка двух десятков тысяч атомов, и это на персональной машине с установленной графической картой со средними по современным меркам характеристиками;
• возможность построения вокруг молекулы триангулированной поверхности типа SES , при этом есть возможность управлять параметрами триангуляционной сетки, а также строить поверхность для больших молекул, таких, как белки;
• возможность изменения ориентации одной молекулы относительно других с использованием оригинального редактора вращений, использующего кватернионную алгебру для определения матриц конечного поворота.
Молекулярный редактор MolRed [4] был создан для визуального наблюдения и редактирования молекулярных структур. Центральной частью программы-редактора является набор инструментов интерактивного взаимодействия пользователя с программой. Этот набор является неотъемлемой частью графического интерфейса программы. От интерактивных возможностей, от удобства их использования во многом зависит привлекательность программы среди пользователей. Программа MolRed , будучи программой-редактором молекулярных структур, прежде всего, должна иметь богатый набор интерактивных инструментов. Более того, именно интерактивные возможности программы и составляют основное её содержание. В данной статье приводится описание основных интерактивных возможностей программы, а также поясняются особенности их реализации.
Следует отметить, что описываемые в статье интерактивные методы широко используют сообщения операционной системы о тех или иных действиях пользователя в окне программы. Использование кросс-платформенной библиотеки QT 4 [5] позволяет отвлечься от учёта особенностей генерирования и передачи приложению сообщений от операционной системы и использовать единый подход в рамках QT 4.
Программа MolRed написана на языке C ++ с использованием объектно-ориентированного программирования, предоставляющего возможности гибкого использования инструментов библиотеки QT 4, которая также написана на C ++. В программе MolRed помимо инкапсуляций данных и методов в классы наиболее широко используется наследование классов от классов библиотеки QT 4, а также полиморфизм перегружаемых в дочерних классах виртуальных функций, обрабатывающих события, поступающие от операционной системы.
2. Типы инструментов взаимодействия с программой
Основные возможности программы были описаны ранее в сообщении [4]. Основные интерактивные элементы программы перечислены ниже:
• Окно графического отображения молекул.
• Окно отображения структур молекул в виде дерева.
• Окно выбора атомов.
• Окно выбора молекул и фрагментов.
• Окно редактирования углов поворота и векторов трансляции отдельных молекул относительно других.
Помимо этих элементов программа также имеет и ряд интерактивных функций, не имеющих отдельного оконного отображения, но вызываемых определёнными действиями пользователя (двойной щелчок, нажатие правой кнопки мыши, клик мышью с одновременным нажатием клавиши Shift и другие). Список основных интерактивных функций:
• Функция навигации в пространстве 3D-сцены графического отображения.
• Функция выделения атома в окне графического отображения.
• Функция выделения молекулы в окне графического отображения.
• Функция выделения набора атомов в окне графического отображения с использованием рамки.
• Функция инвертирования выделения.
• Функция выбора атомов в окне отображения структур.
• Функция определения и отображения метрических параметров выделенных атомов (координаты, длины связей и углы).
• Функция редактирования кратности связей между двумя выделенными атомами
.
• Функция редактирования метрических параметров выделенных атомов (координаты, длины связей, углы).
• Функции разделения молекулы на две части и более, а также объединения двух и более молекул в одну.
• Функции undo и redo для команд пользователя.
Помимо указанных функций имеются функции общего характера, как, например, функция удаления выделенного атома или молекулы, функция записи данных в файл или чтение данных из файла, функция экспортирования текущего изображения 3D-сцены в файл-картинку.
Описанные выше интерактивные элементы и функции определяют базовую интерактивность программы, необходимую для работы с молекулярными структурами.
2.1 Окно графического отображения молекул
 Окно графического отображения молекул реализовано с использованием стандарта OpenGL . При этом использовались возможности библиотеки QT 4 [5], которая инкапсулирует функциональность OpenGL [6, 7] в специальный класс QGLWidget . В программе MolRed создаётся класс OpenGLWidget , являющийся дочерним от QGLWidget . Тем самым класс OpenGLWidget , получает возможность использовать инструменты OpenGL . Инициализация устройства OpenGL осуществляется с помощью инструментов библиотеки QT 4, но при этом параметры инициализации определены в виртуальной перегруженной функции void OpenGLWidget::initializeGL() . В конструкторе класса OpenGLWidget определяются базовые параметры отображения, например, такие как угол перспективы, а также многочисленные второстепенные флаги определяющие режим окна 3D-отображения. Главные методы класса OpenGLWidget - это методы, формирующие 3D-сцену и позволяющие осуществлять навигацию в ней.
Окно графического отображения молекул реализовано с использованием стандарта OpenGL . При этом использовались возможности библиотеки QT 4 [5], которая инкапсулирует функциональность OpenGL [6, 7] в специальный класс QGLWidget . В программе MolRed создаётся класс OpenGLWidget , являющийся дочерним от QGLWidget . Тем самым класс OpenGLWidget , получает возможность использовать инструменты OpenGL . Инициализация устройства OpenGL осуществляется с помощью инструментов библиотеки QT 4, но при этом параметры инициализации определены в виртуальной перегруженной функции void OpenGLWidget::initializeGL() . В конструкторе класса OpenGLWidget определяются базовые параметры отображения, например, такие как угол перспективы, а также многочисленные второстепенные флаги определяющие режим окна 3D-отображения. Главные методы класса OpenGLWidget - это методы, формирующие 3D-сцену и позволяющие осуществлять навигацию в ней.
Формирование 3D-сцены производится с использованием списков объектов. Списки объектов в OpenGL [6, 7] представляют собой структурированные данные, загруженные в память платы графического ускорителя. Поэтому использование списков позволяет как увеличить скорость формирования сцены, так и ускорить сам процесс навигации.
Ниже перечислены методы, формирующие 3D-сцену:
• rendPrmtv(); -формирование базовых примитивов: сферы единичного радиуса, цилиндра единичной длины и единичного радиуса, пары цилиндров и тройки цилиндров. Также здесь формируются изображения строчных символов;
• rendAtoms (); -формирование набора сфер. Положения, радиусы и цвета сфер соответствуют положению, радиусу и типу атомов составляющих отображаемую молекулу;
• rendBonds (); -формирование набора цилиндров, соответствующих связям между атомами;
• rendWire (); -формирование системы соединённых отрезков, соответствующих представлению молекулы в виде решётки;
• rendMol (); -формирование геометрического образа молекулы;
• rendAtCh(); -формирование наборов буквенных символов , соответствующих типам атомов;
• rendScene(); -формирование всех объектов сцены;
• rendSurface (); -формирование молекулярной поверхности.
Формирование базовых примитивов осуществляется с помощью средств OpenGL , но при этом сгенерированные данные, описывающие примитив , запоминаются в списке объектов. На этом этапе генерирование данных для описания примитива происходит с помощью центрального процессора. Но в дальнейшем формирование всех остальных компонент сцены происходит с многократным использованием данных уже сгенерированных и размещённых на микросхемах графической карты, при этом используется процессор графической карты, а не центральный процессор. Такой подход позволяет ускорить формирование объектов 3D-сцены.
Отличие от этой схемы составляет только процедура формирования молекулярной поверхности. Здесь после предварительного расчёта координат точек триангулированной сетки, моделирующей поверхность, создаётся единый объект типа набора объединённых треугольников, который добавляется в список объектов. Формирование поверхности - относительно длительная процедура [8]; основное время связано с расчётом точек молекулярной поверхности. Для построения поверхности используется алгоритм [8], ранее разработанный в нашей лаборатории. Этот алгоритм позволяет строить поверхность типа SES (поверхность исключенного объёма) [9] вокруг выбранной молекулы в виде регулярной триангуляционной сетки как для малых, так и для очень больших молекул. Такие поверхности используются в неявных (континуальных) моделях растворителя, в которых влияние растворителя на свойства молекул учитывается путем заполнения пространства, занимаемого растворителем, например, водой, вокруг молекулы растворяемого вещества, непрерывным диэлектриком с диэлектрической проницаемостью, соответствующей диэлектрической проницаемости растворителя (см., например, [10, 11] и приведенные там ссылки). Для использования, например, метода PCM ( Polarized Continuum Model ) такая поверхность должна быть достаточно гладкой, поскольку именно на ней решаются интегро-дифференциальные уравнения для нахождения поверхностного наведенного заряда. Кроме того, оказалось, что такая поверхность удобна, например, для визуального анализа положений молекул-ингибиторов в активных центрах белков-мишеней.
Несмотря на длительность процедуры построения поверхности для очень больших молекул, навигация в 3D-сцене с отображаемыми молекулами и поверхностями происходит достаточно быстро – насколько это позволяет видеокарта для данного размера молекулярной поверхности и количества точек её триангуляции.
Использование описанного способа построения 3D-сцены с применением списков объектов обеспечивает приемлемую частоту смены кадров даже при работе с очень большими молекулами, содержащими до 20000 атомов.
Функция навигации
Помимо формирования 3D-сцены класс OpenGLWidget также реализует и навигацию в области 3D-сцены. Навигация представляет собой способ изменения относительного расположения и ориентации объектов 3D-сцены и «виртуальной камеры», с помощью которой определяются условия проецирования сцены на экран монитора. Сама процедура проецирования осуществляется методами библиотеки OpenGL . Параметры этого проецирования определяются расположением виртуальной камеры в 3D-сцене. Виртуальная камера может быть представлена как прямоугольная рамка в пространстве 3D-сцены и связанный с ней локальный базис, две оси x и y которого лежат вдоль сторон рамки, а третья ось z определяет направление просмотра в сцене. При этом стороны этой рамки ассоциируются с границами прямоугольной области окна, в котором отображается 3D-сцена. Трёхмерные объекты проецируются на плоскость этой рамки, и результат этого проецирования отображается на экране монитора. Процедуры проецирования реализуются в соответствии со стандартом OpenGL и с помощью его библиотек.
Навигация представлена в виде орбитально – вращательных перемещений виртуальной камеры и поступательных перемещений камеры. При этом:
• Орбитальное движение происходит вокруг неподвижного «центра навигации», который отображается в окне 3D-сцены в виде маленькой сферы. Центр навигации – условная точка в пространстве сцены. Виртуальная камера при любых своих перемещениях всегда направлена (осью z ) на центр навигации. В этом режиме навигации возможно приближение или удаление виртуальной камеры от центра навигации.
• При поступательных перемещениях виртуальной камеры также происходит и перемещение центра навигации, так что относительное расположение камеры и центра навигации не меняются. Перемещения камеры и центра навигации происходят параллельно плоскости рамки виртуальной камеры.
Эти два способа позволяют переместить виртуальную камеру практически в любое место 3D-сцены и получить требуемый ракурс изображения.
Навигационные перемещения орбитального типа происходят при движениях мышью с зажатой левой кнопкой, а поступательные навигационные перемещения происходят при движениях мышью с зажатой правой кнопкой мыши. Для обеспечения такого поведения в классе OpenGLWidget были перегружены соответствующие виртуальные функции-обработчики событий нажатия кнопок мыши, двойных щелчков мыши, а также события перемещения мыши. Все эти события генерируются операционной системой, а средства библиотеки QT 4 [6] обеспечивают связь этих событий с вызовом тех или иных функций-обработчиков. Эта связь уже заложена в реализации базовых классов, которые были наследованы при создании класса OpenGLWidget .
Функции-обработчики нажатия кнопок мыши изменяют внутреннее состояние окна отображения 3D-сцены так, что функция-обработчик движения мыши производит перемещения виртуальной камеры либо в соответствии с орбитальными перемещениями, либо с поступательными. При отпускании кнопки мыши состояние окна возвращается в исходное, когда не производится обработка событий, относящихся к движению мыши.
Поступательные перемещения в 3D-сцене происходят таким образом, что движение мышью вперёд-назад соответствует перемещениям вверх-вниз всей отображаемой картины 3D-сцены. Такое поведение обеспечивается перемещением виртуальной камеры в пространстве 3D-сцены вдоль осей x и y рамки камеры. Одновременно с этим происходит и такое же перемещение центра навигации, так что ось камеры не меняет своего направления. Аналогично перемещения мыши вправо – влево приводят к перемещению всей сцены относительно наблюдателя также вправо – влево. При этом скорость и направление перемещения мыши однозначно пересчитываются в скорость и направление поступательного перемещения виртуальной камеры.
При орбитальной навигации также используются две степени свободы плоского движения мыши. В этом режиме перемещения мыши пересчитываются в соответствующие углы поворота вокруг осей локального базиса, связанного с виртуальной камерой. В пространстве 3D-сцены эти оси меняют направление при орбитальной навигации, но зато для пользователя вращение вокруг этих осей легко интерпретируется и представляется удобным. Здесь движение мышью вперёд-назад соответствует движению вокруг горизонтальной оси, так что ближайшие к виртуальной камере фрагменты сцены прокручиваются вверх-вниз. Движение мышью влево - вправо соответствует движению вокруг вертикальной оси и аналогичным прокручиваниям влево - вправо.
Отметим, что перемещения и повороты сцены, которые осуществляются в режиме навигации, не соответствуют изменениям координат объектов сцены. Все объекты при этом не изменяют своего положения. Изменяет своё положение и ориентацию только виртуальная камера.
Функция выделения атома
Выделение атома в программе производиться двойным щелчком левой кнопки мыши по атому. При этом вызывается функция-обработчик события двойного щелчка. Данная функция после успешной проверки состояния окна 3D-сцены (не включен ли один из дополнительных режимов, как, например, режим редактирования ориентации молекулы) запускает процедуру определения номера атома, который выбрал пользователь. Процедура определения выбранного атома использует специальный режим проецирования из библиотеки OpenGL , который позволяет выделить список объектов, проецирующихся в заданную область экрана. В этой процедуре выбирается маленькая область экрана: всего 2х2 пикселя, расположенная под курсором мыши. Далее запускается обработка сцены, состоящей только из атомов, и с помощью OpenGL функции
glRenderMode(GL_RENDER) определяются номера объектов, которые проецируются в указанную область экрана. Из всех объектов выбирается самый ближайший к виртуальной камере. В конце определятся номер молекулы, которой принадлежит атом и номер этого атома в молекуле. В соответсвующем поле структуры, описывающей молекулу, запоминается номер выделенного атома. При построении 3D-сцены данные о выделенных атомах используются для изменения цвета отображения атома.
Если выделение производится с удержанрием клавиши Shift , то выделенный атом добавляется к выделенным ранее, если же выбран был ранее выделенный атом, то выделение этого атома снимается.
Функция выделения молекулы
Эта функция работает аналогично функции выделения атома, с тем исключением, что после определения номера молекулы, которой принадлежит отмеченный пользователем атом, все атомы данной молекулы отмечаются как выделенные.
Функция выделения рамкой группы атомов
Функция, также аналогичная функции выделения одного атома, но с тем отличием, что область проецирования выбирается не 2х2, а в соответсвии с размером рамки, заданной пользователем, и, кроме этого, выделяется не один ближайший к виртуальной камере атом, а все атомы, которые проецируются внутрь области, очерченой рамкой.
Функция инвертирования выделения
Вызывается отдельной кнопкой на панели инструментов. Производит инвертирование выделения, так что те атомы, которые были выделены, перестают быть выделенными, а невыделенные атомы помечаются, как выделенные. Эта функция работает как в режиме выделения атомов, так и в режиме выделения молекул.
Функции определения метрических и угловых параметров выделенных атомов
Эта функция анализирует количество выделенных атомов и в зависимости от их числа определяет координаты одного атома, расстояние между двумя атомами, валентный угол между тремя атомами и торсионный угол, определяемый четырмя атомами. Эти значения отображаются в нижней части главного окна программы. Определение метрических параметров производится на основе декартовых координат атомов. Расстояние между двумя атомами определяется как модуль вектора разности координат двух атомов. Углы определяются с использованием скалярных (для торсионного угла – ещё и векторных) произведений векторов разности координат двух атомов.
Функция редактирования метрических параметров
При выделении одного, двух, трёх или четырёх атомов возможно редактировать соответсвующий метрический или угловой параметр. Для выделенного одного атома возможно его перемещние вдоль плоскости виртуальной камеры. Для этого двойным щелчком правой кнопки мыши программа переводится в режим поступательных смещений выделеных атомов. Повторный двойной щелчок возвращает программу в исходный режим. Редактирование длины связи производится при помощи удержания клавиши Shift , нажатия левой кнопки мыши на один из выделеных атомов и движением мыши. В этом режиме изменяется только длина связи, но не меняется её ориентация. В режиме редактирования длины связи вектор перемещения мыши преобразуется в вектор перемещения атома вдоль прямой, соединяющей его с другим выделенным атомом, таким образом атом перемещается вслед за курсором мыши. Аналогично возможно редактировать значения валентого и торсионного углов. При этом мышь должна наводиться либо на первый, либо на последний из выделенных атомов. При редактировании валентного угла остаётся неизменной плоскость валентного угла, а при редактировании торсионного угла – прямая между вторым и третьим выделенными атомами. Здесь также применяются методы преобразования векторов смещения мыши в соответсвующие изменения значений углов, так, чтобы перемещеамый атом отслеживал изменения курсора мыши.
Функция изменения кратности связи
Позволяет изменить кратность связи при выдедении двух атомов в одной молекуле. Кратность связей может быть одинарной, двойной и тройной. Также эта функция позволяет удалить связь. Функция вызывается нажатием на соответсвующую кнопку на панели инструментов. Связь может быть добавлена при выделении двух атомов одной молекулы. Если выделенные два атома принадлежат разным молекулам, то вызов этой функции блокируется.
2.2 Окно отображения структур молекул в виде дерева
 Окно отображения структур молекул в виде дерева реализовано с использованием концепции «модель/представление», реализованной в библиотеке QT 4. В рамках этого подхода достаточно реализовать структуру для хранения отображаемых данных каждого элемента (атома, молекулы), и использовать эту структуру для создания структуры данных в виде дерева (модель) и структуры отображения (представление) на основе предоставляемой библиотекой QT 4 «заготовок». В программе Molred создан специальный класс TreeItem , описывающий отображаемые данные каждой молекулы и каждого атома. Данный класс соответствует типу узла дерева. Ещё один класс TreeModel , наследованный от класса QAbstractItemModel библиотеки QT 4, реализует струкруру данных в виде дерева (модель), узлами которого являются объекты типа TreeItem . И ещё один класс TreeView , наследуемый от QTreeView библиотеки QT 4, определяет отображение дерева в графическом окне (представление), а также описывает реакцию на определённые действия пользователя, то есть определяет интерактивное поведение окна отбражения структур молекул в виде дерева.
Окно отображения структур молекул в виде дерева реализовано с использованием концепции «модель/представление», реализованной в библиотеке QT 4. В рамках этого подхода достаточно реализовать структуру для хранения отображаемых данных каждого элемента (атома, молекулы), и использовать эту структуру для создания структуры данных в виде дерева (модель) и структуры отображения (представление) на основе предоставляемой библиотекой QT 4 «заготовок». В программе Molred создан специальный класс TreeItem , описывающий отображаемые данные каждой молекулы и каждого атома. Данный класс соответствует типу узла дерева. Ещё один класс TreeModel , наследованный от класса QAbstractItemModel библиотеки QT 4, реализует струкруру данных в виде дерева (модель), узлами которого являются объекты типа TreeItem . И ещё один класс TreeView , наследуемый от QTreeView библиотеки QT 4, определяет отображение дерева в графическом окне (представление), а также описывает реакцию на определённые действия пользователя, то есть определяет интерактивное поведение окна отбражения структур молекул в виде дерева.
Основные обрабываемые действия пользователя в области описываемого окна - это вызов контекстного меню и двойной щелчок левой кнопкой мыши. Содержание контекстного меню описывется в рамках класса TreeView, и оно зависит от типа выделенного элемента, например, для атома или группы атомов контекстное меню оличается от меню для молекулы. В контекстном меню пользователь может выбрать то или ное действие, например, выделить атом на 3D-сцене, или изменить тип отображения молекулы. Двойной щелчок мыши просто приводит к снятию выделения ранне отмеченных элементов в окне отбражения структур молекул в виде дерева (но не к снятию выделения атомов отмеченных на 3D-сцене).
В окне древовидного отображения могут быть представлены не только данные о молекулах и их атомах, но и о других объектах сцены, таких, как молекулярная поверхность. При этом также возможны выделения и вызов контекстного меню с пунктами, соответствующими действиям с поверхностью (поменять цвет повехности, изменить тип отображения поверхности).
Функция выделения атомов с помощью окна отображения структур молекул в виде дерева
Выделение атома осуществляется с помощью вызова контексного меню и выбора пункта выделения атома. Перед вызовом контекстного меню нужно пометить один или несколько атомов. Также возможно и выделение молекулы - для этого нужно пометить пункт, соотвествующий молекуле, и вызвать контексное меню, в ктором выбрать пункт выделения молекулы в 3D-сцене. Возможность выделения атомов и молекул с помощью окна древовидного отображения является дополнительным способом выделения и позволяет выбрать атомы, например, по их номеру.
2.3 Окно выбора атомов
 Окно выбора атомов реализовано в виде всплывающего окна отдельного от окна основной программы. Внешний вид окна представляет собой таблицу Менделеева химических элементов, где каждому элементу соответствует кнопка. Пользователь выбирает тип атома кликом по кнопке-атому, а затем добавляет атом выбранного типа в область 3D-сцены. Для выхода из режима добавления атомов достаточно закрыть окно или произвести двойной щелчок правой кнопкой мыши по области 3D-сцены.
Окно выбора атомов реализовано в виде всплывающего окна отдельного от окна основной программы. Внешний вид окна представляет собой таблицу Менделеева химических элементов, где каждому элементу соответствует кнопка. Пользователь выбирает тип атома кликом по кнопке-атому, а затем добавляет атом выбранного типа в область 3D-сцены. Для выхода из режима добавления атомов достаточно закрыть окно или произвести двойной щелчок правой кнопкой мыши по области 3D-сцены.
Реализация окна выбора атомов (виртуальная таблица Менделеева) выполнена в виде класса MendeleevTable , который наследован от класса QDialog библиотеки QT 4, описывающего диалоговое окно. Для удобства окно виртуальной таблицы Менделеева имеет статус отображения поверх других окон. В состав класса MendeleevTable добавлен массив кнопок, каждая из которых ассоциирована с определённым типом атома. Тип атома отображается на кнопке в виде символов химического элемента. Каждая кнопка является объектом класса QPushButton , который определён также в библиотеке QT 4 и реализует собой кнопку. В конструкторе класса MendeleevTable производится создание массива кнопок, ассоциация каждой кнопки с типом атома и размещение кнопок в области виртуальной таблицы Менделеева в соответствии с общепринятым расположением элементов в периодической таблице. Кроме этого, с помощью системы сигналов библиотеки QT 4 сигнал, имитируемый каждой кнопкой при клике по ней мышью, связан с методом (процедурой) анализа клика. В этой процедуре производится идентификация нажатой кнопки в массиве всех кнопок, запоминает номер кнопки и изменение шрифта текста, отображающего тип химического элемента этой кнопки, на более жирный и если ранее уже был выбран какой-либо атом, то шрифт его кнопки возвращается на обычный. По окончании работы процедуры анализа испускается ещё один сигнал, который связан с классом OpenGLWidget отображающем 3D-сцену. Класс OpenGLWidget изменяет при этом своё состояние и переходит в режим добавления атомов. В этом режиме клики мышью по области окна 3D-сцены обрабатываются не так, как при режиме навигации.
Двойной щелчок мыши по пустому месту в окне 3D-сцены приводит к добавлению атома выбранного ранее типа в месте, где был произведён клик мышью. При этом применяется процедура восстановления трёхмерных координат по имеющимся двумерным координатам положения мыши относительно экрана. Процедура вычисляет точку в 3D-пространстве, которая лежит «под» курсором мыши в плоскости, параллельной плоскости экрана, и проходит через центр навигации. Вычисленные координаты присваиваются новому атому. В этом режиме помимо того, что в 3D-сцену добавляется атом, на уровне структуры описания молекул также добавляется новая молекула, состоящая из одного вновь добавленного атома. То есть новые атомы добавляются без связи с уже имеющимися в 3D-сцене молекулами, а как новые отдельные молекулы, состоящие из одного атома.
При двойном щелчке по атому из 3D-сцены тип этого атома заменится на тип нового атома. При этом «новый» атом будет автоматически включён в состав исходной молекулы и будет иметь те же координаты, что и его предшественник.
Ещё один способ добавить атом – это добавить его вместе со связью к какой-либо молекуле. Для этого производится одиночный клик по существующему атому, а затем ещё один такой же клик по тому месту, где предполагается поместить новый атом. При этом новый атом оказывается связанным одинарной связью с первоначально отмеченным атомом. В этом режиме также используется процедура вычисления 3D-координат, но в отличие от ранее описанной процедуры здесь плоскость вычисляемой точки, оставаясь параллельной экрану, проходит не через центр навигации, а через первоначально отмеченный атом.
Описанные способы добавления атомов позволяют добавлять как одиночные атомы, так и атомы, связанные с существующими молекулами, а также производить замену типа атома.
2.4 Окно выбора молекул
 Окно реализовано в виде класса FragmentBrowser , который включает в себя два различных окна: окно 3D-сцены для отображения выбираемой молекулы( ViewScene ), и окно древовидного списка названий молекул ( FragmentList ), которые можно выбрать и добавить в основную рабочую область. Эти два окна реализованы аналогично описанному выше окну графического отображения молекул и окну отображения структур молекул в виде дерева. Само окно выбора молекул отображается отдельно от основного окна программы и имеет статус отображения поверх других окон.
Окно реализовано в виде класса FragmentBrowser , который включает в себя два различных окна: окно 3D-сцены для отображения выбираемой молекулы( ViewScene ), и окно древовидного списка названий молекул ( FragmentList ), которые можно выбрать и добавить в основную рабочую область. Эти два окна реализованы аналогично описанному выше окну графического отображения молекул и окну отображения структур молекул в виде дерева. Само окно выбора молекул отображается отдельно от основного окна программы и имеет статус отображения поверх других окон.
Класс FragmentList создаёт древовидный список названий молекул, причём структура дерева списка соответсвует расположению файлов с описанием молекул на жёстком диске. В конструкторе этого класса происходит чтение содержимого специальной папки с именем ‘ Structures ', в которой хранятся файлы с данными о молекулах и заполнением списка названий молекул. В конструкторе класса FragmentBrowser происходит связывание сигнала клика мыши в окна древовидного списка с методом определения выбранной молекулы и отображения её в окне графического отбражения класса ViewScene . При первоначальной загрузке окно графического отображения пусто. При выборе молекулы из списка имён в окне появляется её геометричческая структура. Интерактивные возможжности окна ViewScene представлены орбитатьной навигацией, то есть выбираемую молекулу можно рассмотреть с разных ракурсов, приблизить или отдалить. Центр навигации при этом помещён в центр молекулы и не отображается на экране.
После выбора молекулы пользователь может выбрать в пространстве основной 3D-сцены место, куда нужно добавить молекулу, и двойным щелчком левой кнопки мыши добавить выбранную молекулу. При этом ориентация новой молекулы будет соответсвовать той, в которой она записана в исходном файле. Новая молекула добавляется в массив молекул рабочей сцены без образования каких-либо связей со старыми молекулами, даже при возможном частичном геометричеком пересечении с ними.
2.5 Окно редактирования углов поворота и векторов трансляции отдельных молекул
 Данное окно соответствует переходу в режим поворота и трансляции одной выделенной молекулы относительно глобальной системы координат. Отметим, что вращение молекулы в этом режиме отличается от вращений, которые наблюдаются пользователем при поворотах сцены в режиме навигации. В случае навигации вращается виртуальная камера, а молекулы остаются на месте, и их координаты в глобальной системе не меняются. Реализация окна выполнена в виде класса RotatWin , наследующего класс QDialog библиотеки QT 4. В состав класса входят параметры, определяющие угол поворота и смещение выделенной молекулы, единичный вектор оси поворота и вектор положения центра оси, а также значения инкремента угла поворота и смещения. Смещение производится вдоль оси вращения. При вызове окна класса RotatWin центр смещения помещается в геометрический центр молекулы. Начальные значения поворота и смещения равны нулю.
Данное окно соответствует переходу в режим поворота и трансляции одной выделенной молекулы относительно глобальной системы координат. Отметим, что вращение молекулы в этом режиме отличается от вращений, которые наблюдаются пользователем при поворотах сцены в режиме навигации. В случае навигации вращается виртуальная камера, а молекулы остаются на месте, и их координаты в глобальной системе не меняются. Реализация окна выполнена в виде класса RotatWin , наследующего класс QDialog библиотеки QT 4. В состав класса входят параметры, определяющие угол поворота и смещение выделенной молекулы, единичный вектор оси поворота и вектор положения центра оси, а также значения инкремента угла поворота и смещения. Смещение производится вдоль оси вращения. При вызове окна класса RotatWin центр смещения помещается в геометрический центр молекулы. Начальные значения поворота и смещения равны нулю.
Вызов окна редактирования ориентации молекул включает соответствующий режим окна 3D-сцены. В этом режиме при удержании клавиш Shift и Ctrl осуществляется редактирование ориентации выделенной молекулы.
Shift + левая кнопка + движение мышью приводит к непреывному изменению оринтации оси вращения. Изменение ориентации сразу отображается в окне 3D-сцены, а тажке меняются координаты оси, отображаемые в окне класса RotatWin . Ориентацию оси поворота можно задать и вручную, указав в окне редактирования ориентации координы оси вращения.
Shift + правая кнопка + движение мышью приводит к непрерывному перемещению центра оси вращения в плоскости, паралельной плоскости виртуальной камеры (паралельно плоскости экрана). Здесь также отображаются координаты центра оси в окне класса RotatWin , и также можно задать положения центра оси вручную, указав координаты.
Shift +клик левой кнопкой по атому приводит к изменению оси вращения в направлении из текущего центра оси на выбранный атом.
Shift + клик правой кнопкой по атому приводит к переносу центра оси на выбранный атом, направление оси при этом не меняется.
Shift + вращение колёсика мыши приводит к изменению величины угла поворота вокруг оси вращения. Изменение угла вращения происходит на величину инкремента угла, знак изменения зависит от направления вращения колёсика мыши. По умолчанию угол инкремента составляет 0.05 радиан. Угол поворота, как и значение инкремента, можно задать вручную в соотвествующих полях окна класса RotatWin . При изменении угла поворота вычисляется новое положение молекулы и отображается на экране.
Ctrl +вращение колёсика мыши приводит к изменению смещения молекулы вдоль оси вращения. Изменение смещения происходит также на величину инкремента, знак зависит от направления вращения колёсика мыши. По умолчанию инкремент составляет 0.05 ангстрем. Значение инкремента, как и значение самого смещения, можно задать вручную.
Нажатие на клавишу Enter приводит к фиксированию произведённого поворота и смещения, дальнейшие повороты и смещения будут отсчитываться от текущего положения молекулы. Выход из режима редактирования ориентации молекулы производится двойным щечком правой кнопки мыши.
Синхронизация между действиями пользователя по редактированию ориентации молекулы и отображением как самой молекулы, так и параметров поворота в окне класса RotatWin, осуществляется с помощью системы сигналов библиотеки QT 4. При этом действия пользователя по редактированию углов поворота и смещений могут проявляться как через действия мышью, так и через явное задание координат. В первом случае сигналы о изменении геомертии исходят из класса OpenGLWidget и передаются главному управляющему модулю программы, откуда посылаются к классу RotatWin для отображения новых значений параметров. Во втором случае, наоборот, сигнал об изменении геометрии испускается классом RotatWin , передаётся главному управляющему модулю и оттуда передаётся модулю изменения геометрии молекулы. Класс OpenGLWidget автоматически воспроизводит изменённую геометрию молекулы.
Изменения ориентации молекулы производятся на основе вычиления матрицы поворота и последующего её применения для координат каждого атома. Для вычисления мартицы поворота на заданный угол вокруг заданной оси используются матричные представления нормированных кватернионов. Как известно[12-14], кватернионы представляют собой четвёрку чисел q ={ q0 , q1 , q2 , q3 } , с особыми правилами умножения. В векторном виде кватернионы записываются в виде:
q = q0 + q1i + q2j + q3k , где q0, q1, q2, q3 - вещественные числа, и введены орты (базисные векторы) i , j и k , для которых введены правила умножения: ii = jj = kk = ijk = -1; ij = k , ji = - k .
Помимо векторной формы существует и матричная форма записи кватерниона, при этом различают два типа матриц: левая и правая.
Для произведения двух кватернионов a и b : c = ab = Ab = Ba , при этом A – матричное левостороннее представление кватерниона a , B –матричное правостороннее представление кватерниона b . Связь матричных представлений с компонентами кватерниона следующая:
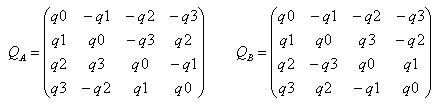
Произведение 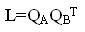 при условии нормировки
при условии нормировки 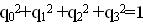 даёт ортогональную матрицу, которая поворачивает векторную часть кватерниона ( q1 , q2 , q3). Для матрицы L справедливо представление:
даёт ортогональную матрицу, которая поворачивает векторную часть кватерниона ( q1 , q2 , q3). Для матрицы L справедливо представление:
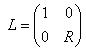 ,
,
где R – матрица поворота в трёхметрном пространстве вокруг оси, определяемой векторной частью кватерниона на угол, определяемый его скалярной частью. Аналогично формуле Эйлера для комплексных чисел кватернионы представляются в виде:
q = cos(?/2)+ nsin (?/2) = exp( n?/2) , при этом n – единичный веткор направления векторной чатси кватрениона, а угол ? – угол поворота вокруг оси n .
Исходя из требуемых направлений n и угла ? , можно построить кватернион в обоих матричных формах и получить соответствующую этому направлению и углу матрицу поворота R .
2.6 Функция разделения и объединения молекул
На уровне логического описания молекул и атомов, принятого в программе, наименьшей самостоятельной единицей представления данных является молекула. То есть в программе не может быть описан некоторый атом, который не входил бы в состав молекулы. Поэтому даже для описания отдельного атома, не связанного ни с какими другими атомами, создаётся отдельная структура описывающая молекулу, которая состоит из этого одного атома. Например, при добавлении несвязанных атомов в сцену с помощью виртуальной таблицы Менделеева, на каждый новый атом создаётся новая структура, описывающая молекулу с одним атомом. Если требуется, например, создать связь между двумя такими отдельными атомами, или иными двумя молекулами, то их необходимо объединить в одну молекулу. Эту задачу выполняет функция объединения выделенных молекул в одну молекулу. При этом все атомы исходных молекул оказываются логически объединены в одну молекулу. В рамках этой новой молекулы, например, доступна функция добавления связей. Такое объединение также полезно использовать и для других операций, когда несколько молекул необходимо обработать как единое целое, например, повернуть относительно некоторой оси, или построить общую молекулярную поверхность.
Обратная функция - разделение молекулы на отдельные молекулы. Функция проводит анализ связности атомов, входящих в молекулу, и выделяет наборы связанных атомов. В рамках одного такого набора атомов каждый атом связан с любым другим атомом через последовательность промежуточных связей атомов, входящих в этот набор. И в тоже время ни один атом из одного набора не может быть связан ни с каким атомом другого набора. На основе таких наборов связанных атомов формируются новые молекулы.
Поиск связанных наборов атомов осуществляется с помощью специальной рекурсивно вызываемой функции. Функция вызывается для каждого атома и опрашивает соседей атома: не вызывалась ли эта функция для них ранее. Если атом не обрабатывался, то производится рекурсивный вызов функции для этого нового атома. Процесс оканчивается, когда не остаётся необработанных атомов, связанных с уже обработанными. Таким образом, все обработанные атомы входят в состав одной группы связанных атомов. Процесс повторяется сначала, пока не будут обработаны все атомы молекулы. При этом формируются наборы связанных между атомов.
2.7 Функции “ undo ” и “ redo ” для команд пользователя
Эти функции считаются неотъемлемой частью любого редактора. Они позволяют отменить или вернуть действие пользователя над объектом редактирования. В программе MolRed эта функциональность реализована с помощью класса undoredo , в рамках которого принята условная шифровка команд пользователя и установлена привязка определённого типа данных к каждой команде. Каждая команда означает некоторое действие по изменению состоянию программы, используя при этом соответствующие данные, привязанные к команде. Кодируются и отслеживаются не все действия пользователя, а только значимые, например, навигационные действия пользователя программа не отслеживает, а действия по перемещению атома или молекулы – отслеживает. Программа не запоминает полное состояние объектов редактирования, а лишь последовательность команд и соответствующих каждой команде данных. Команды хранятся в линейном массиве, при этом используется индикатор текущей команды. Данные для каждого типа команд также хранятся в массивах. При этом для каждой команды указывается номер ячейки из массива данных, где лежат данные для этой команды. Для каждой команды имеется обратная команда. Например, команда удаления атома имеет обратную команду – добавления атома. Исключение составляют команды смещений – обратимость действия команды легко осуществить, если изменить знак смещения на обратный.
Пользователь может вызвать действие undo комбинацией клавиш Ctrl + Z и действие redo комбинацией Ctrl + Y , или соответствующими кнопками на панели инструментов.
При поступлении новой команды из числа отслеживаемых программа запоминает новую команду в наборе команд, формирует соответствующие этой команде данные, которые также запоминаются. После этого производится выполнение этой новой команды. Выполнение команды увеличивает индикатор текущей команды на единицу. При этом в конце исполнения команда в своей ячейке массива команд меняет себя на обратную и также меняет данные команды на соответствующие обратной команде.
Действия redo и undo осуществляются исполнением команд с меньшим или с большим номером команды в массиве команд относительно индикатора текущего положения. Для действия undo исполняется команда с номером на единицу меньшим, чем текущее положение индикатора, а для действия redo – на единицу большим, если, конечно, текущее положение индикатора не является последним. Если положение индикатора соответствует последней команде, то действие redo невозможно, и программа блокирует вызов этого действия. Изменение команды на обратную автоматически гарантирует выполнение обратных операции при движении по массиву команд в сторону более ранних команд, и выполнение прямых команд при движении в сторону более поздних команд.
Примеры использования интерактивных возможностей редактора Molred приведены ниже на рисунках.
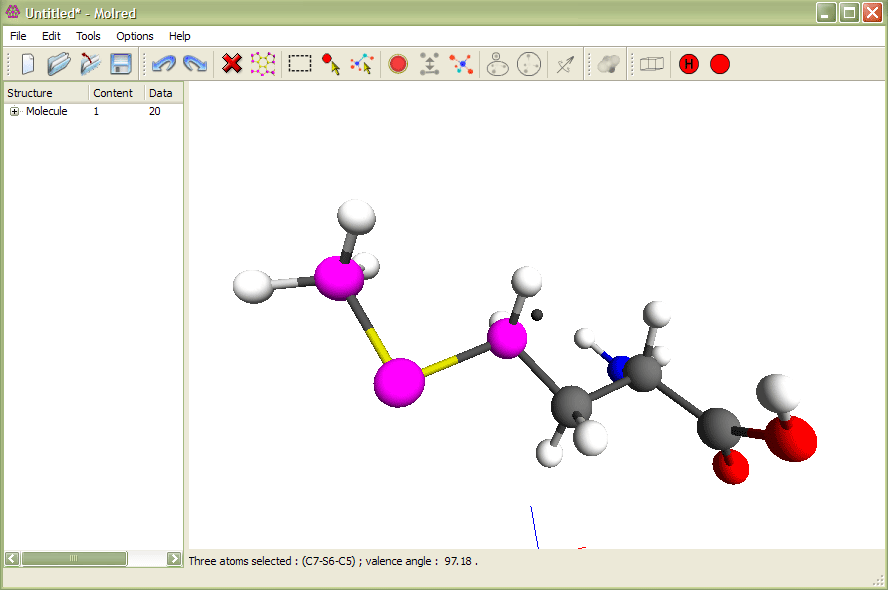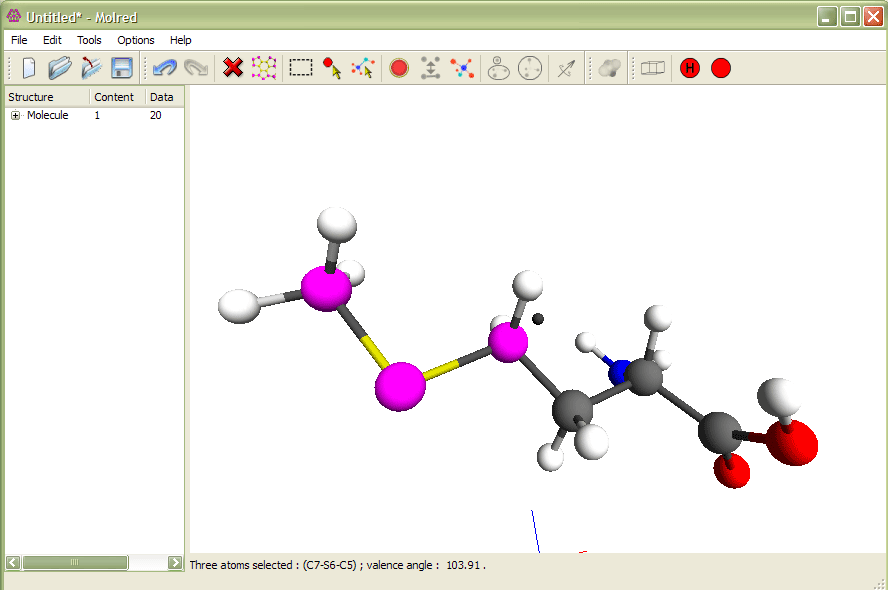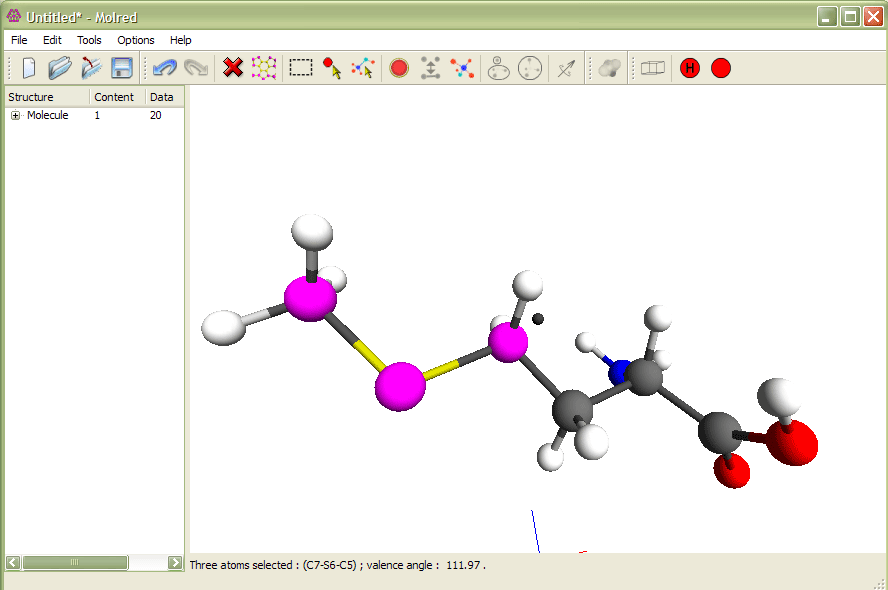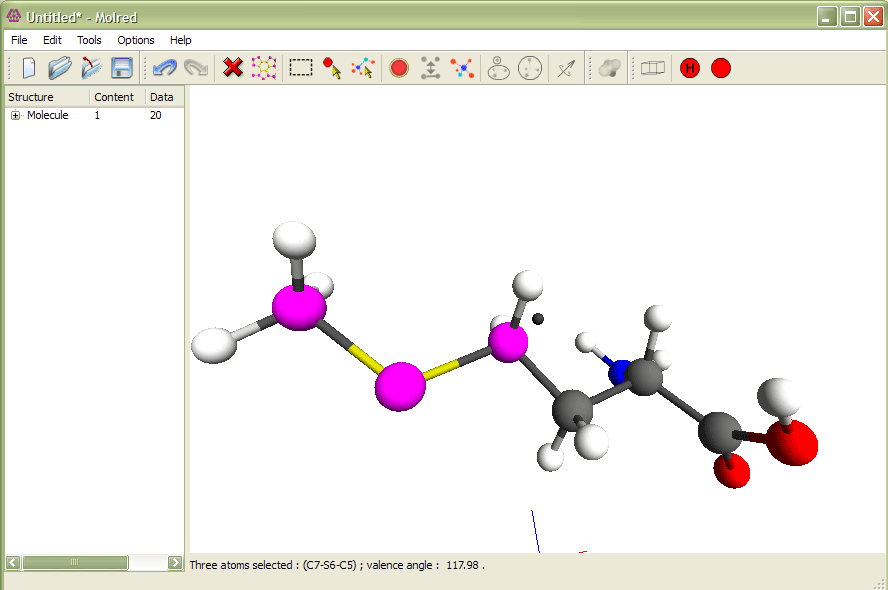
Рис.1
Выделение трех атомов и изменение валентного угла. Анимация иллюстрирует процесс изменения валентного угла.
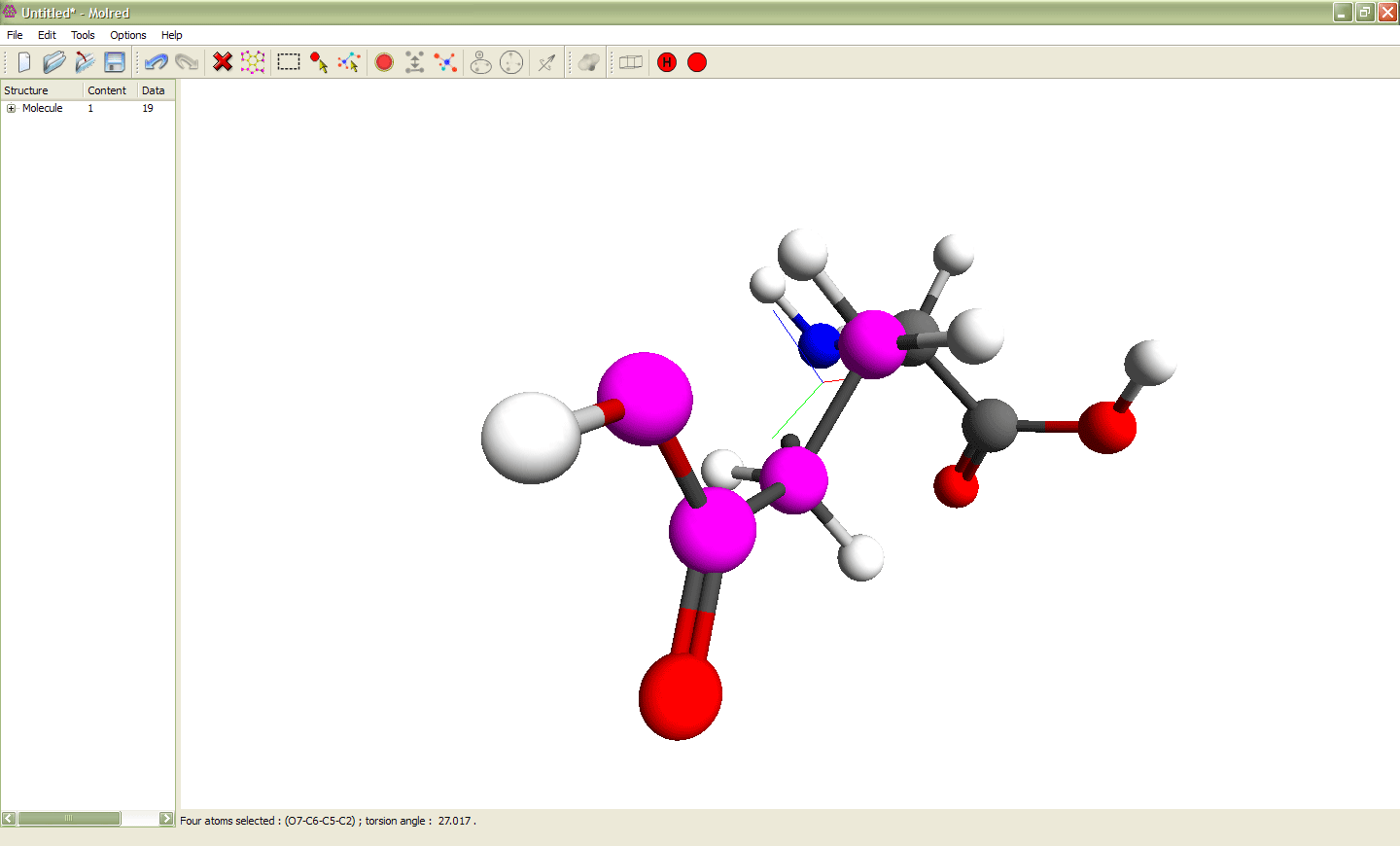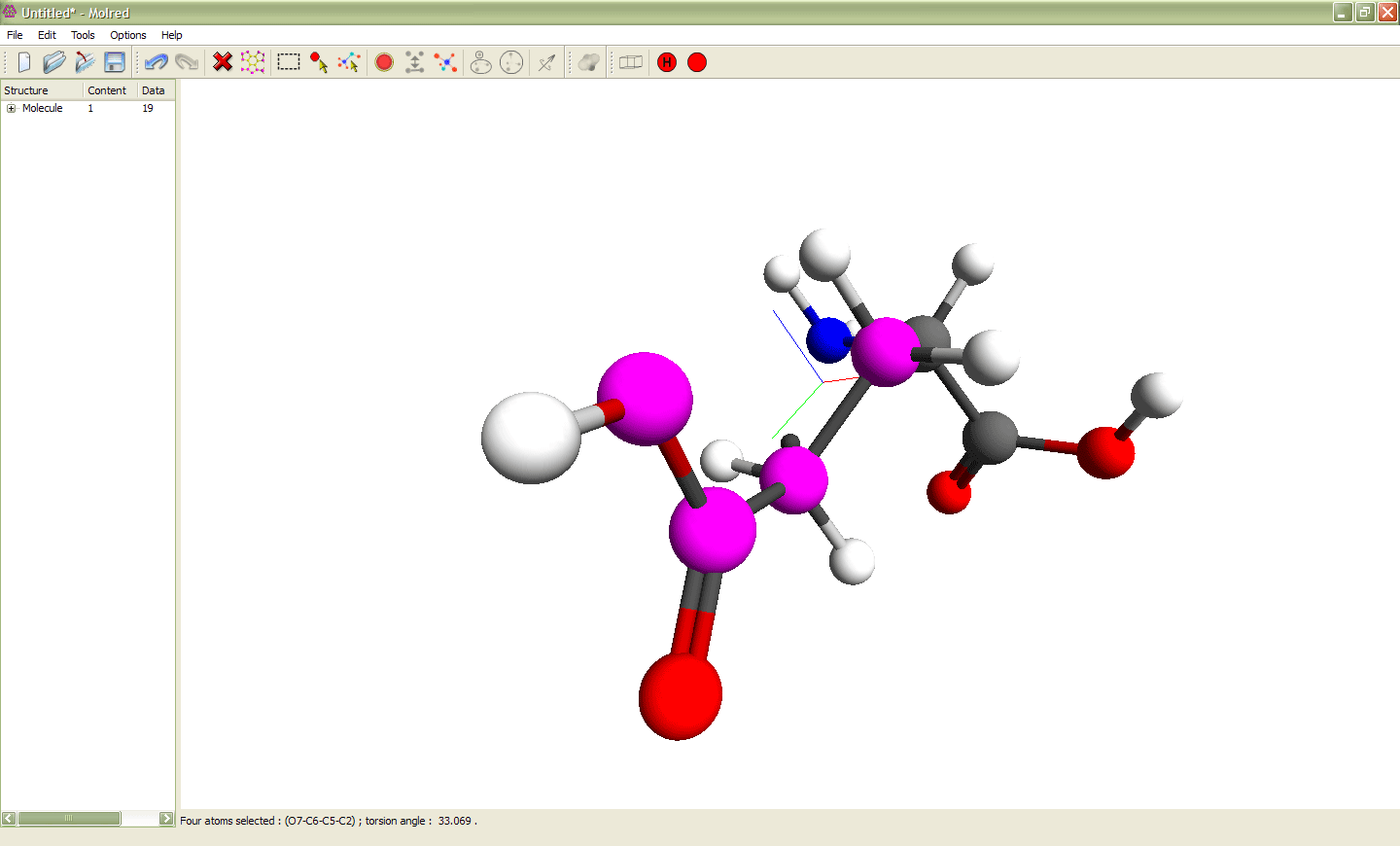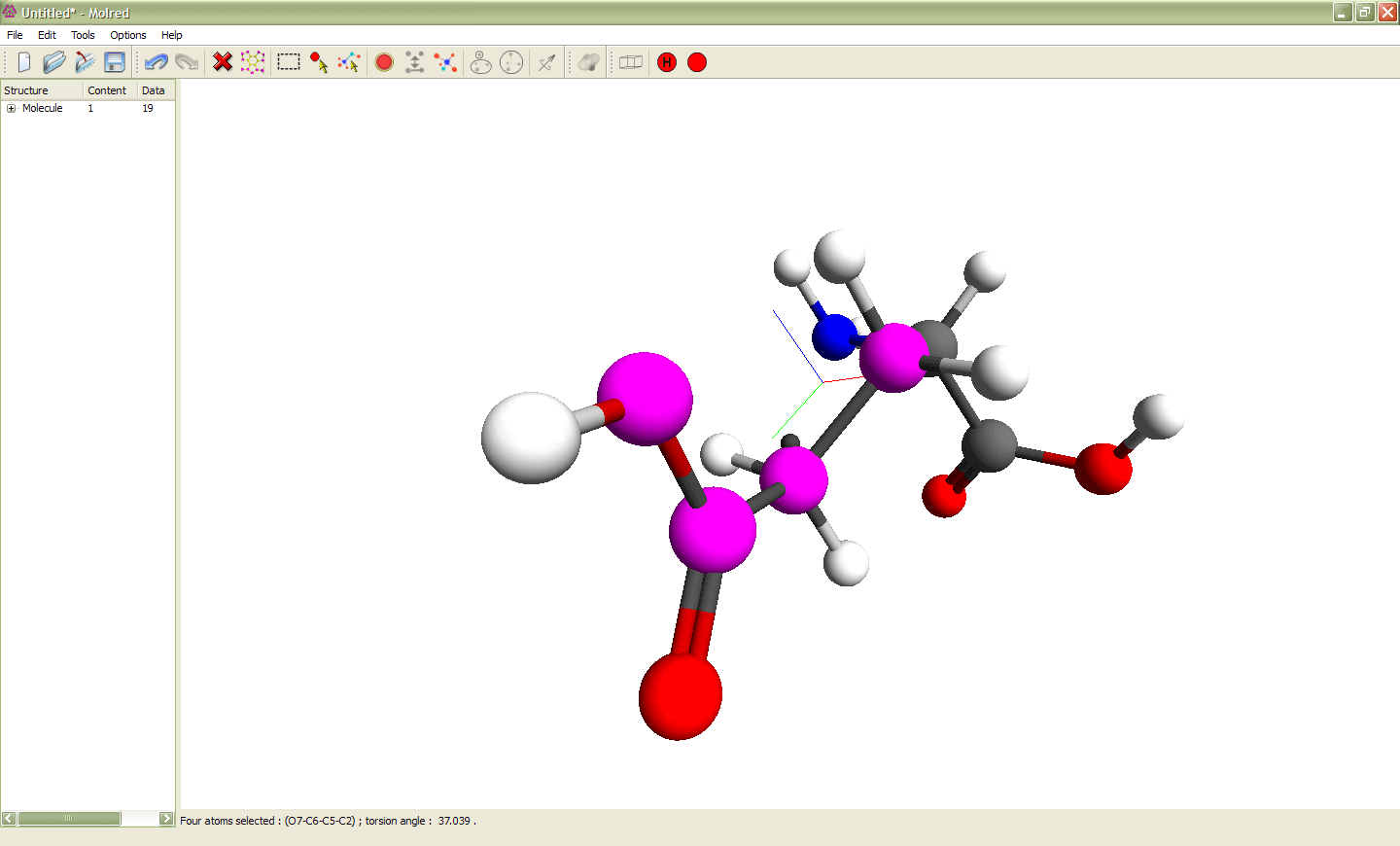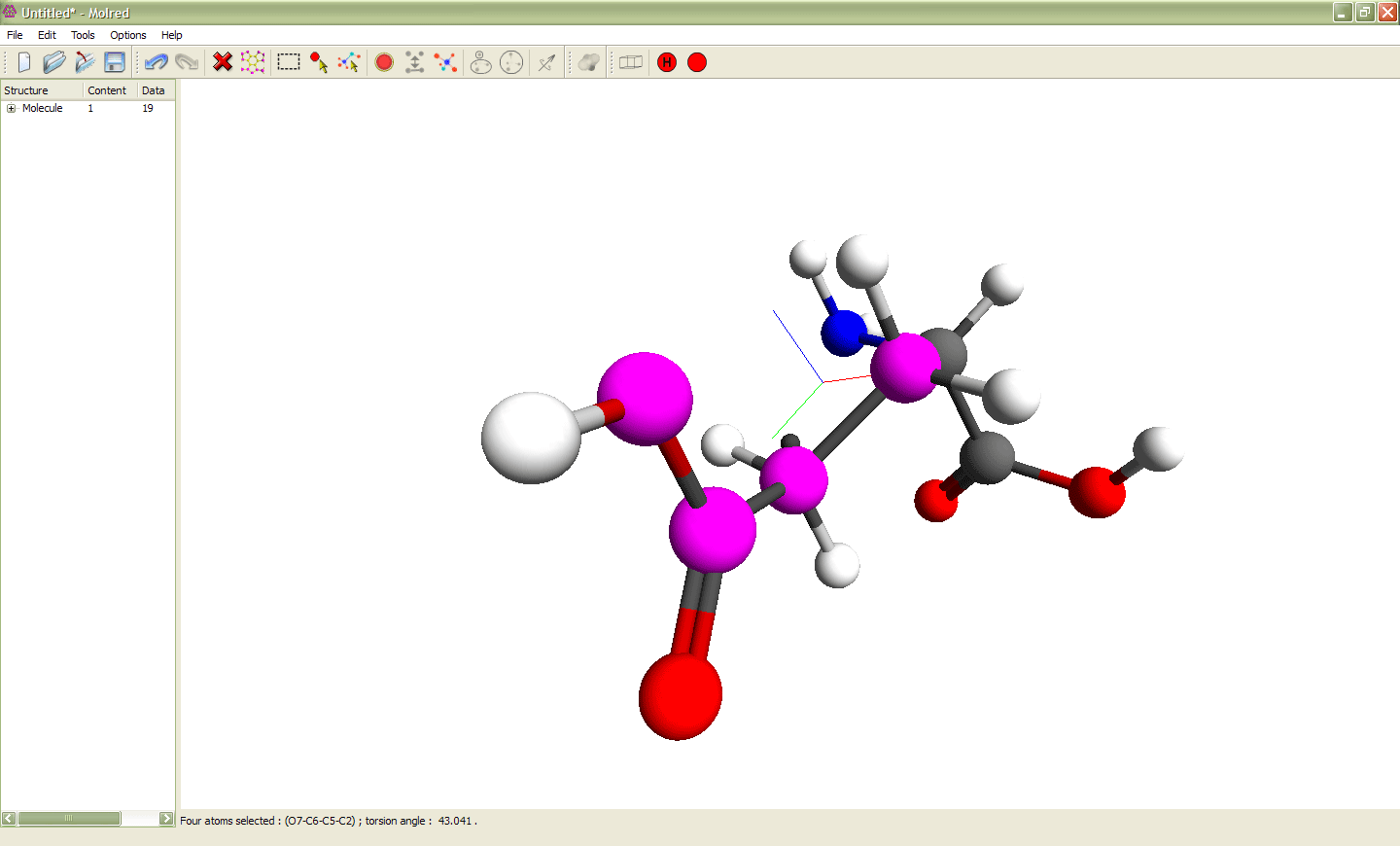
Рис. 2
Выделение 4 атомов и изменение торсионного угла ими определяемого. Выделенные атомы показаны сиреневым цветом. Эти атомы образуют торсионный угол, значение которого можно изменять. Анимация иллюстрирует процесс изменения торсионного угла.
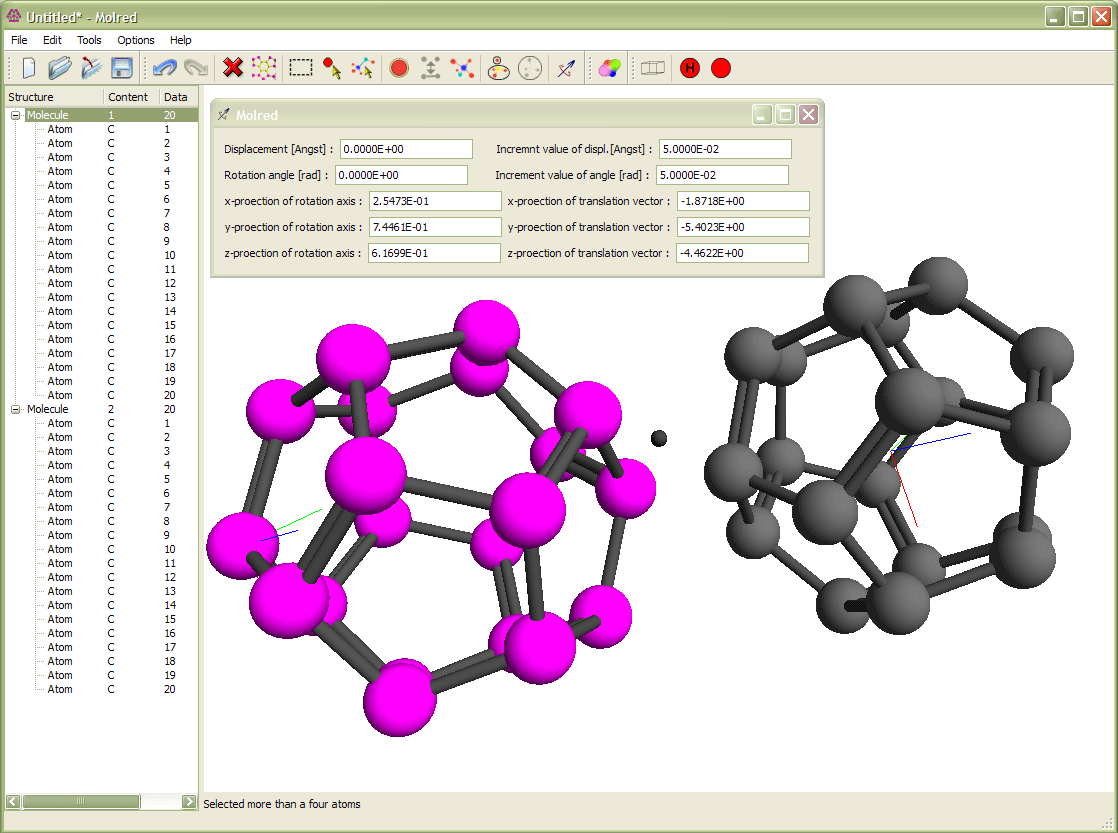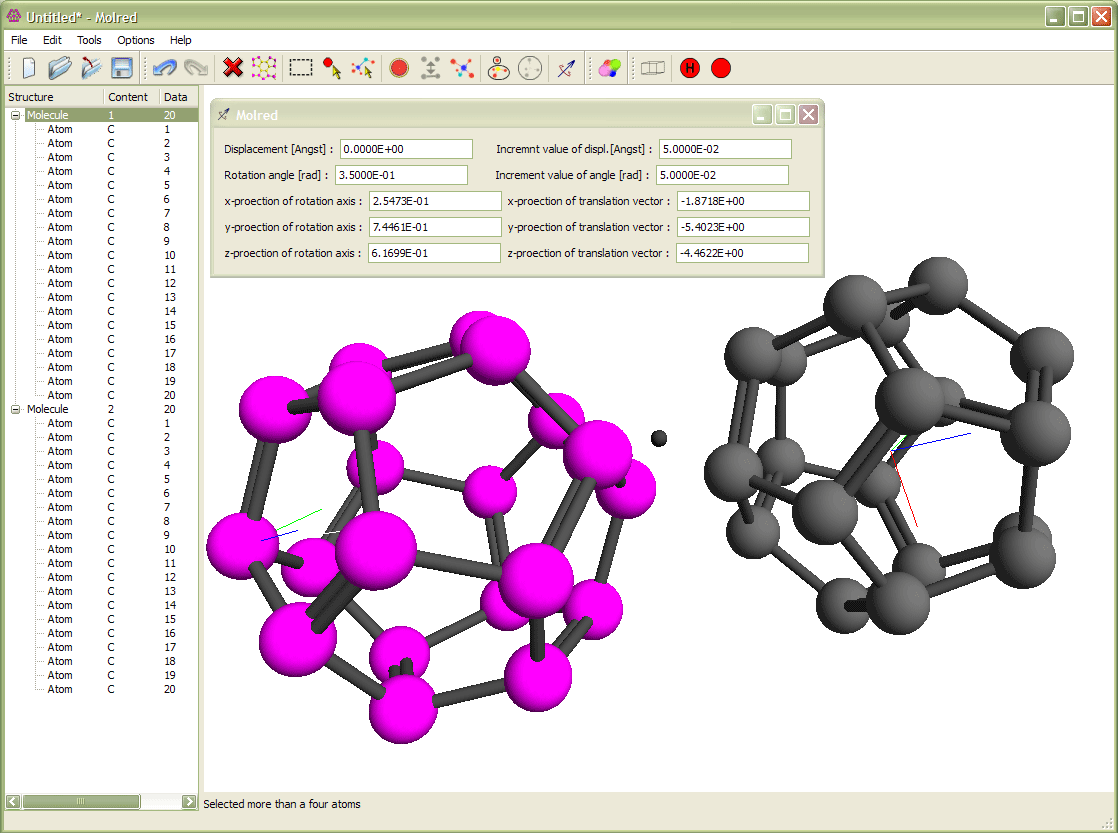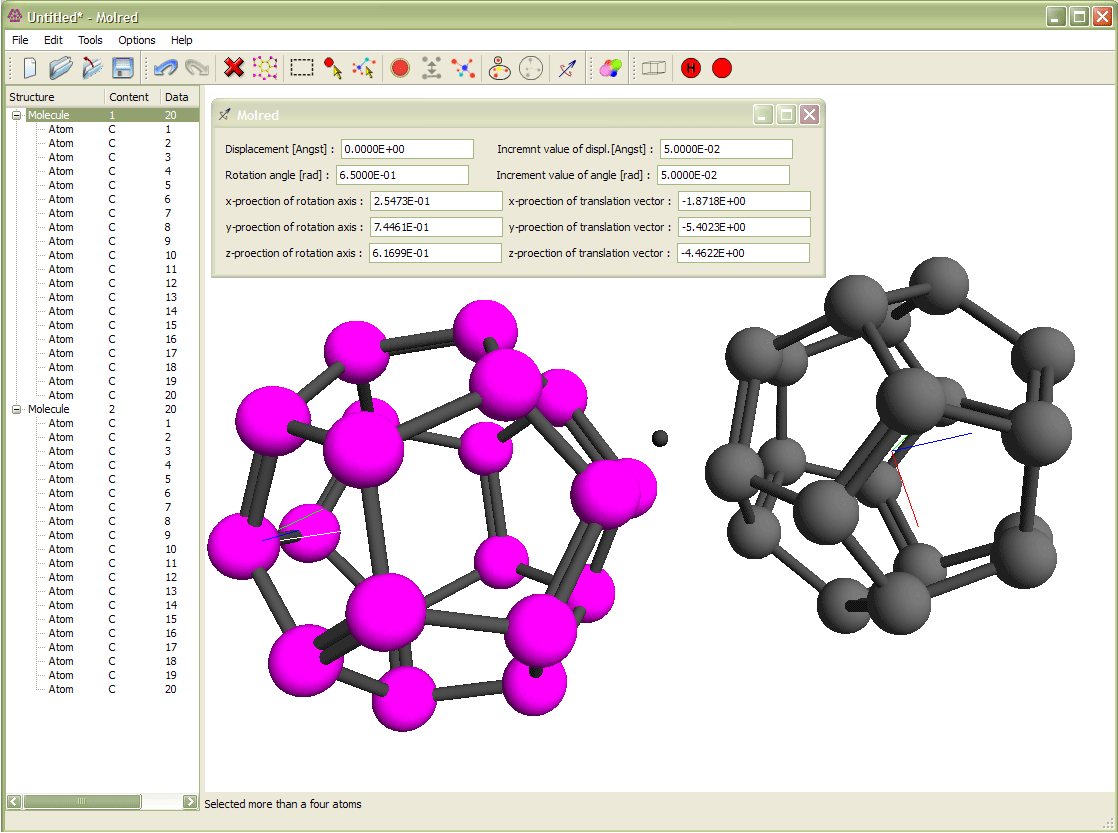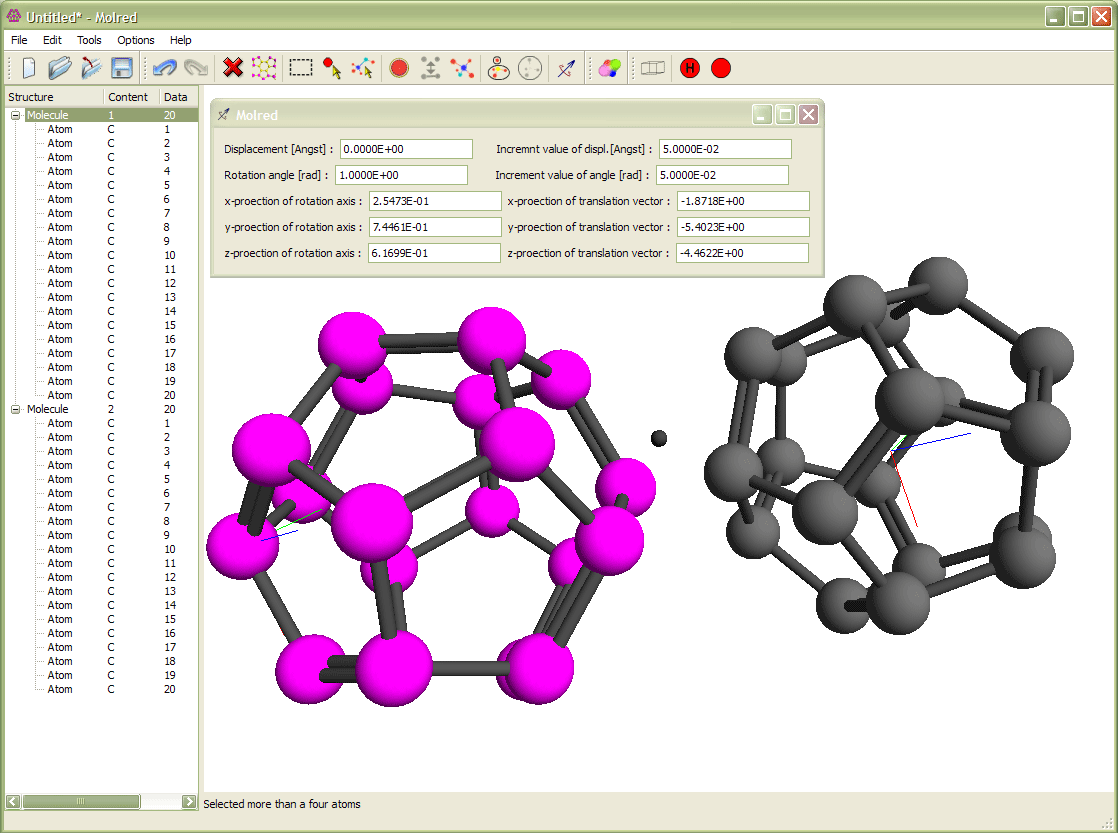
Рис. 3
Выделение молекулы и её вращение относительно другой молекулы. Вращение происходит вокруг оси с направлением, выбранным от одного атома молекулы к другому. Анимация иллюстрирует процесс поворота молекулы.
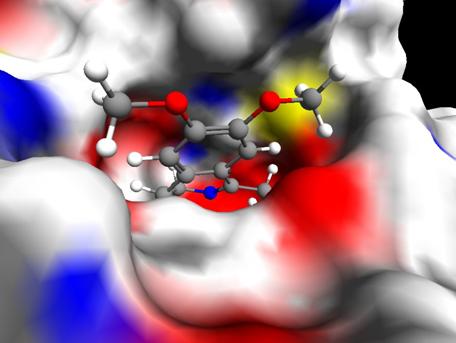
Рис. 4
На рисунке показан ингибитор, находящийся в активном центре белка - урокиназы. Подавление протеолитической функции этого белка ведет к заметному уменьшению процесса метастазирования клеток злокачественных опухолей. Имеются также сведения о том, что урокиназа играет важную роль в процессах роста и распространения опухолей. Однако надлежащее функционирование урокиназы необходимо для осуществления процесса ангиогенеза, например, урокиназа способствует процессам рубцевания после инфаркта . Всё это делает урокиназу привлекательной мишенью для разработки новых лекарств на основе её ингибиторов.
На приведенном выше рисунке вокруг урокиназы построена поверхность исключенного объёма, а молекула-ингибитор представлена в модели шариков и цилиндров. Положение ингибитора в активном центре урокиназы получено с помощью программы докинга SOL [15] в процессе разработки новых ингибиторов урокиназы. Цвета поверхности соответствуют типам атомов, находящихся вблизи данного участка поверхности: красный – кислород, синий – азот, желтый – сера, белый – водород, серый – углерод. Такое представление делает удобным анализ взаимного расположения молекулы-ингибитора и белка, и, тем самым, дает эффективный инструмент разработчикам новых лекарств, поскольку основой большинства современных лекарств являются ингибиторы белков-мишеней, определяющих развитие заболевания.
3. Заключение
В работе описаны основные интерактивные возможности программы MolRed . Указаны методы реализации этих возможностей и способы вызова функций редактора пользователем. Отметим широкое использование в программе библиотеки OpenGL для формирования трёхмерных сцен отображения геометрических структур и библиотеки QT 4 для формирования оконного графического интерфейса. Рассмотренные в настоящей работе интерактивные возможности позволяют уже сейчас широко использовать данный молекулярный редактор для создания, просмотра, редактирования и анализа молекулярных структур, в том числе применять его для разработки новых лекарств.
Авторы благодарны Федору Григорьеву, Анне Головачевой, Алексею Сулимову, Виталию Авакяну, Ксении Владимировой, а также профессору Александру Багатурьянцу за полезные обсуждения и ценные замечания.
Работа выполнена при поддержке грантов РФФИ 09-01-12097 офи-м, 09-07-12135 офи-м, Госконтрактов между Федеральным агентством по науке и инновациям и НИВЦ МГУ имени М.В.Ломоносова № 02.435.11.1008 от 01.08.2005 и № 02.514.11.4084 от 08.08.2008, а также контракта между Центром Фотохимии РАН и ООО «Димонта» № 02.523.11.3014-ДМ/08 от 25.08.2008.
Список литературы
- В.А.Садовничий, В.Б.Сулимов Суперкомпьютерные технологии в медицине, в Сборнике «Суперкомпьютерные технологии в науке, образовании и промышленности», Под редакцией В.А.Садовничего, Г.И.Савина и Вл.В.Воеводина, Издательство Московского Университета, Москва, 2009, с. 16-23.
- http://www.camd.ru/visualization.html
- http://avogadro.openmolecules.net/
- Жабин С.Н., Сулимов В.Б. “Программа для визуализации и редактирования молекул « Molred »”, Электронный журнал «Научная визуализация», Национальный Исследовательский Ядерный Университет "МИФИ", 2009, Т.1 №1, с.108-114.
- http://www.qtsoftware.com/
- http://www.opengl.org/
- http://www.glprogramming.com/red/
- Жабин С.Н., Сулимов В.Б. “Программа построения доступной растворителю поверхности для произвольных органических молекул и интерактивный просмотр положений лигандов в активных центрах белков”, Сборник материалов XIII российского национального конгресса «Человек и лекарство», 3-5 апреля 2006, стр.15.
- M. L. Connolly. "Analytical molecular surface calculation." J Appl Cryst, 16(5):548–558, Oct 1983
- Tomasi, J.; Mennucci, B.; Cammi, R. Molecules in solution. In Handbook of Molecular Physics and Quantum Chemistry ; Wilson , S., Ed.;Wiley: Chichester , U.K. , 2003; p 299.
- Romanov A.N., Jabin S.N., Martynov Y.B., Sulimov A.V., Grigoriev F.V., Sulimov V.B. “ Surface Generalized Born method: a simple, fast and precise implicit solvent model beyond the Coulomb approximation ”, J.Phys.Chem. A, 108, N 43 (2004) 9323-9327
- W.R. Hamilton. Elements of Quaternions, volume I. Chelsea Publishing Company, third edition, 1969.
- On quaternions and octonions: Their geometry, arithmetic, and symmetry , by John H. Conway and Derek A. Smith, A K Peters, Ltd., Natick, MA, 2003, xii + 159 pp
- http://zhurnal.ape.relarn.ru/articles/2002/033.pdf
- Романов А.Н., Кондакова О.А., Григорьев Ф.В., Сулимов А.В., Лущекина С.В., Мартынов Я.Б., Сулимов В.Б. Компьютерная разработка лекарств: программа докинга SOL // Вычислительные методы и программирование, 2008, Т. 8, С.213-233.